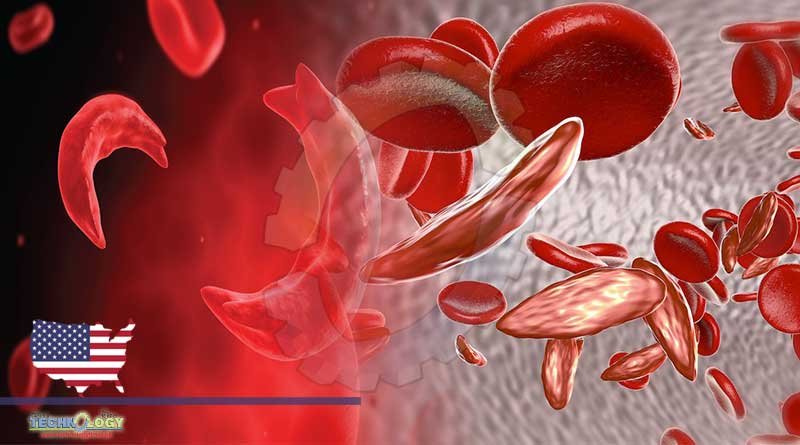
A January study published found CRISPR, FDA-approved, could forge new treatment for curing blood disorders such as sickle cell disease.

Gene editing has upended many areas of science, from creating pesticide-free food to attempting to bring back the wooly mammoth. Using a technology called CRISPR-CAS9, commonly referred to as CRISPR, scientists are now trying to fix genetic errors that cause disease.
A January study published in The New England Journal of Medicine found CRISPR could forge new treatments for curing blood disorders such as sickle cell disease.
Patients diagnosed with sickle cell disease have a mutation in a gene for hemoglobin—an iron-rich protein in red blood cells. The mutation causes abnormally C-shaped blood cells, which have a hard time carrying oxygen to other parts of the body.2 Its hard and sticky feature also clogs blood flow, which increases the risk for infections.
Gene Therapy for Sickle Cell: Looking for a Disease Cure
Sickle cell disease is an inherited blood disorder that affects about 100,000 Americans per year, Alexis A. Thompson, MD, MPH, former president of the American Society of Hematology and pediatric hematologist at the Feinberg School of Medicine at Northwestern University, tells Verywell. While Thompson, who was not involved with the study, says children appear relatively normal at birth, it isn’t until 6 to 12 months of age that children start developing problems.
“In the younger age group, patients who present with pain, severe fever, or infection require hospitalization, receiving very strong medication and missing out on school,” Thompson says. “As they move to adulthood, they have challenges finishing education, university, or maintaining a job.” Using CRISPR, researchers are trying to change some of these outcomes.
What This Means For You
Sickle cell disease is passed on to a child when both parents have the sickle cell trait. If you’re unsure of your carrier status, it’s essential to get screened by a healthcare professional. If you do have sickle cell disease, new treatments using CRISPR technology may be available to you in the future.
Genetic Strategy Restarts Hemoglobin Production
The study followed one patient with sickle cell disease and one patient with beta-thalassemia, a blood disorder that reduces the production of hemoglobin.3
Both patients required blood stem cells, but the study sought to use their cells rather than cells from a sibling. When blood stem cells were taken from the patient, the researchers used CRISPR, which acts as a molecular scissor, and a single-guide RNA molecule, CAS9, to locate a specific gene called BCL11A.
The Truth Behind Common Misconceptions of Sickle Cell Disease
In this study, the researchers cut BCL11A because it acts like a genetic switch that turns off the gene that produces a fetal form of hemoglobin. By turning it back on, scientists reactivated the production of fetal hemoglobin, which replaced missing or defective hemoglobin in both patients’ red blood cells. Any remaining diseased cells were eliminated via chemotherapy.
Hemoglobin Levels Remained Stable Months After Treatment
Six and 12 months after the procedure, both patients underwent bone marrow aspirates to measure the number of red blood cells present in their bone sample.
The first patient was a 19-year-old female diagnosed with beta-thalassemia. Four months after her last bone marrow transplant with the gene-edited stem cells, her hemoglobin levels stabilized and remained stable at her last follow-up visit. Although she initially experienced serious side effects from the treatment (pneumonia and liver disease), they resolved after a few weeks.
Why Should I Care About Sickle Cell Trait If It Isn’t a Disease?
The second patient was a 33-year-old female with sickle cell disease. Fifteen months post-procedure, her fetal hemoglobin levels rose from 9.1% to 43.2%. Her mutated hemoglobin levels from sickle cell disease decreased from 74.1% to 52.3%. While she experienced three severe side effects (sepsis, cholelithiasis, and abdominal pain), they were resolved with treatment.
One of the major advantages of this approach, compared to traditional forms of treating these blood disorders, is its use of patient’s cells with no need for a donor.
“Cells of the same patient can be manipulated and can be transplanted without the risk of rejection or to cause immune reactions from the donor (graft-versus-host disease),” Damiano Rondelli, MD, the Michael Reese Professor of Hematology at the University of Illinois at Chicago College of Medicine, said in a statement.4
Since publication, researchers have extended their work to eight more patients—six with beta-thalassemia and three with sickle cell disease. Their current results are consistent with the first two patients in the study.
Current Treatment for Sickle Cell Disease
The current FDA-approved treatment for sickle cell disease is a bone marrow transplant. However, this procedure requires that the patient have a sibling whose tissue perfectly matches theirs.
Thompson says a major treatment challenge is that one in four siblings aren’t the same tissue type. Even if the bone marrow transplant takes place, there are also serious side-effects to the procedure, including graft failure, graft versus host disease, and death.
If bone marrow transplants are out of the picture, an alternative treatment is a haploid identical transplant. “There’s been a success with haploid identical transplants where the tissue type is partially matched, but the transplant is performed in a very different way to achieve an engraftment with your complications,” Thompson says. However, she says only a minority of patients qualify for this treatment.
Red Cross Seeking Black Blood Donors for Critical Shortages
Because of the restrictions and limitations for sickle cell disease, Thompson says that there had been some discussion about having patients serve as their own donors. In this current study, the authors look at gene editing as a potential avenue for this type of treatment.
How Genetic Treatments Can Help
Anyone can inherit sickle cell disease, but it’s especially common in:5
- People of African descent, including African-Americans
- Hispanic-Americans from Central and South America
- People of Middle Eastern, Asian, Indian, and Mediterranean descent
- In the U.S., all children born in the country are screened for sickle cell disease, giving ample opportunity for early treatment. But several scenarios make it challenging to diagnose every case. Thompson says families that immigrated to the U.S. may have older children who have not been screened along with parents who are unaware of their carrier status until they have a child who has the condition.
Learn About the Blood Disorders That the Newborn Screen Can Detect
Despite the imperfections with screenings, industrialized countries have improved their prognosis for sickle cell disease. “Today, a child born today in the United States has a 95% chance of surviving into adulthood, and the same is true for other resourceful countries like the United Kingdom,” Thompson says.
From a global perspective, however, Thompson says low- and middle-income countries might not offer the same treatments that are currently available for people in countries like the U.S. She says over half of children with sickle cell disease in Sub-Saharan Africa will not live beyond their fifth birthday.
Based on the study results, gene editing could help treatments for sickle cell disease become more widely accessible.
“The hope is that this treatment will be accessible and affordable in many low-middle-income countries, the Middle East, Africa, and India, and have an important impact in the lives of many people in these areas,” Rondelli said
Originally published at Very well health