Scientists at the NEI have developed CRX gene therapy strategy that a promises for a rare disease that causes severe vision loss in childhood.

Scientists at the National Eye Institute (NEI) have developed a promising gene therapy strategy for a rare disease that causes severe vision loss in childhood. A form of Leber congenital amaurosis, the disease is caused by autosomal-dominant mutations in the CRX gene, which are challenging to treat with gene therapy.
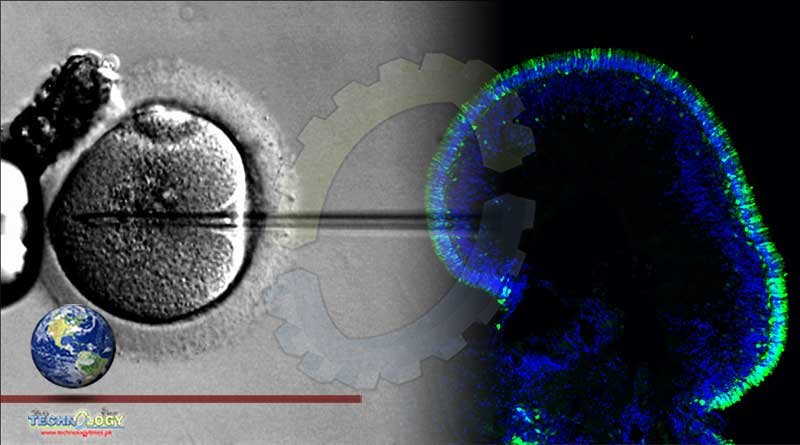
The scientists tested their approach using lab-made retinal tissues built from patient cells, called retinal organoids. This approach, which involved adding copies of the normal gene under its native control mechanism, partially restored CRX function. The study report appears today in Stem Cell Reports. NEI is part of the National Institutes of Health.
The U.S. Food and Drug Administration approved Luxturna in 2017 for the treatment of LCA patients with mutations in a gene called RPE65. Although hailed as a major advance in gene therapy, Luxturna is ineffective against other forms of LCA, including those caused by autosomal-dominant mutations in CRX.
The CRX gene encodes a protein (also called CRX) that binds to DNA and instructs the retina’s photoreceptors to make light-sensitive pigments called opsins. Without functional CRX protein, photoreceptors lose their ability to detect light and eventually die.
Disorders like autosomal-dominant LCA are tricky to treat with gene therapy, because adding more of the normal gene does not always restore function. People with autosomal-dominant mutations still have one normal copy of the gene, but the mutant version of the protein interferes with the normal protein. Sometimes, instead of restoring normal function, simply adding more of the normal protein can enhance the disease in unpredictable ways.
To explore how gene augmentation – adding copies of the normal gene – would affect autosomal-dominant LCA, Swaroop’s team, developed retinal organoids from two volunteers with LCA and from their unaffected family members. Led by Kamil Kruczek, Ph.D., a postdoctoral fellow in Swaroop’s lab, they built the complex retina-like tissues in several stages, starting with skin cells, inducing the production of mature photoreceptors and other retinal cells with the genetic profile of each volunteer. As expected, patient organoids made far less light-sensing opsin than the organoids made from unaffected family members.
To carefully control how much CRX gene would be expressed by the recipient photoreceptors, the team re-engineered the CRXpromoter so it could be delivered with the CRX gene as part of the gene therapy. A promoter is a neighboring sequence of DNA that controls when and how genes are expressed. The researchers packed the gene and their engineered promoter inside a virus that shuttled them into the organoid photoreceptors.
The team’s gene augmentation strategy restored some CRX protein function for organoids from both patients, driving expression of opsins in both types of photoreceptors: rods and cones.
“The fact that this strategy worked for both CRX mutations was pretty exciting,” said Swaroop. “Gene augmentation may be a viable therapy for LCA caused by other autosomal-dominant mutations.”
“This proof-of-concept gene therapy study is the first step toward a potential treatment for a rare form of LCA,” said Brian Brooks, M.D., NEI clinical director and co-author on the study. “It’s a great example of bench-to-bedside science, when researchers in basic and clinical science collaborate.”
Originally published at News Medical